Project 3
Sodium chloride as promoter for vascular disease
Johannes Stegbauer (HHU), Brant Isakson (UVA)
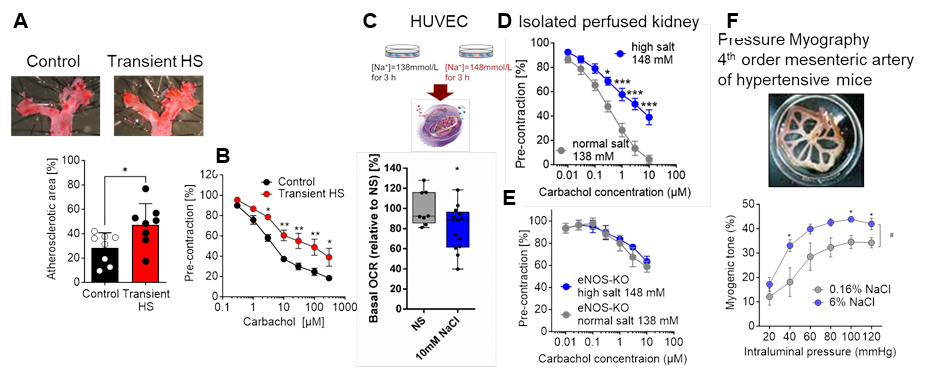
Figure legend: Transient high salt (HS) treatment accelerates atherosclerosis (A) and endothelial dysfunction (B) in aortas of AngII-infused apoE-deficient mice. Acute increase in NaCl reduces mitochondrial basal oxygen capacity rate and impairs renal endothelial function induced by carbachol. In eNOS-KO mice the negative effect of high salt is abolished. Chronic high salt intake (6%) impairs myogenic tone in isolated mesenteric arteries of AngII-infused mice compared to normal salt (0,16% NaCl) treated mice (in cooperation with Isakson lab). *, **, ***p<0.05, 0,01, 0.001.
Background and preliminary work: Continuous progressive changes in vascular structure and function play an important role in the development of vascular disease. The switch from a healthy to a diseased vessel is highly dependent on exogenous and endogenous factors causing changes in the plasticity of endothelial cells (EC) and vascular smooth muscle cells 1, 2. This process is facilitated and characterized by low-grade vascular inflammation, reduced nitric oxide (NO) and mitochondrial dysfunction, consequently causing endothelial dysfunction, extracellular matrix remodeling and increased arterial stiffness 3-6. Beside classical endogenous factors like angiotensin, aldosterone, and pro-inflammatory cytokines, micronutrients such as sodium seem to play an important role for vascular function and plasticity 7-9. Human studies revealed that high salt (NaCl) intake is associated with increased cardiovascular morbidity and mortality 10. Although salt affects blood pressure, the detrimental effects of high salt intake on the vasculature affecting endothelial function and integrity cannot be explained only by an increase in blood pressure. In fact, high salt intake shifted immune cells to a pro-inflammatory phenotype 8, 11. In addition, we and others have shown that high salt impairs mitochondrial function, reduces NO bioavailability, and increases reactive oxidative species (ROS). Since high salt also leads to stiffening of EC 12 and increases myogenic tone in the resistance arteries (Fig. 3), it appears that salt influences the phenotype and plasticity of ECs and promotes progressive vascular damage. Interestingly, it has been proposed that high salt induces some kind of “salt memory”, which is responsible for an aggravated cellular response after a second hypertensive hit 13. In this regard, we have shown that transient high salt treatment followed by a washout period with normal salt intake exacerbates subsequent hypertensive vascular damage such as atherosclerosis and aortic aneurysms (Fig.) suggesting that high salt intake induces a long-term plastic alteration of the vessel and therefore makes it susceptible to vascular damage. However, the exact extent and mechanism of how increased salt intake affects endothelial plasticity and thereby promotes the development of vascular disease is not known.
Hypothesis: Sodium chloride promotes vascular injury by influencing endothelial cell function and plasticity.
Work program: WP1 Impact of an acute high salt load on endothelial function (untargeted and targeted approach) Preliminary results from our lab showed that an acute increase in sodium concentration impairs mitochondrial function and induces endothelial dysfunction by reducing NO bioavailability as this effect was absent in endothelial specific eNOS deficient KO mice (Fig. 3 B-C). Within this aim we will investigate the underlying mechanism how high-salt induces mitochondrial dysfunction and thereby impairs endothelial function in different vascular beds. First, we will analyze whether and how an increase in high salt affects mitochondrial function. Using an untargeted approach, we will measure ATP production, mitochondrial respiration and metabolic pathways as well as intermediates of the TCA and glycolysis by using Seahorse assays, high-resolution respirometry and pulsed stable isotope-resolved metabolomics in primary endothelial cells from resistance (e.g., mesenteric) and conductance (e.g., aorta) arteries incubated with high salt (NaCl 148mmol/l) or normal salt (NaCl 138mmol/l). We will also analyze NADPH oxidase-derived superoxide anion radicals and H2O2 together with NO generation by eNOS using chemiluminescence and electron paramagnetic resonance (EPR) techniques. Moreover, the effects of an acute NaCl increase on endothelial dysfunction in resistance and conductance arteries such as mesenteric, intrarenal, carotid arteries and aortas will be studied by pressure and wire myography as well as the isolated perfused kidney model. Finally we will investigate whether inhibition of ROS production or an improvement of mitochondrial respiration attenuates high-salt induced endothelial function ex vivo and thereby explains how acute salt impairs endothelial function.
WP2 Impact of high salt intake on endothelial plasticity as a promotor for vascular disease (non-targeted approach) Chronic high salt treatment leads to endothelial inflammation, dysfunction, and stiffening all features known to affect endothelial plasticity. Within this aim, we will investigate how chronic high salt intake modulates endothelial function and plasticity in ECs. Furthermore, we will analyze, whether these structural and functional effects are still observed after stopping high salt treatment and are thus responsible for an accelerated vascular damage after a second hit (aim 3). We hypothesize that high salt treatment leads to long-lasting changes in endothelial plasticity after discontinuation of high salt intake. For this reason, we will first investigate how chronic salt intake (6 % NaCl for 6 weeks) affects endothelial function and myogenic tone in the same murine vessels as described in aim 1. Further, to investigate whether salt-induced endothelial dysfunction is reversible, we will perform the same vascular measurements two weeks after stopping salt intake. To gain a deeper and unbiased insight how salt affects endothelial cells plasticity and to determine whether these effects are still evident after salt treatment has stopped, we will perform bulk RNAseq analysis of isolated ECs from these different arteries by using YFP-Cdh5-CreERT2+ mice. In this case, bulkRNAseq provides a much deeper read of the RNA than scRNAseq could provide, including miRNAs. In addition, using the YFP-tagged endothelium will greatly enhance endothelial purity and viability when these specific arteries are used. Using these same arteries, we will also perform ATACseq to determine at the transcription level whether the “salt memory” has been activated. With help of this unbiased bioinformatic approach, we will elucidate specific effects of salt intake on the different vascular systems. Furthermore, we will discover signaling pathways that are responsible for endothelial dysfunction, inflammation and proliferation and compare them with the results obtained in WP1. Finally, we will identify transcriptome signatures that may not revert after discontinuation of salt and therefore make the endothelium more susceptible to vascular damage.
WP3 Alterations in endothelial plasticity caused by transient high salt intake exacerbate hypertensive vascular damage (non-targeted approach) In this aim, performed during the exchange phase in the Isakson lab at UVA, we will investigate whether changes in endothelial plasticity induced by high salt aggravates hypertension induced vascular dysfunction. Therefore, hypertensive vascular damage will be induced in mice after transient high salt intake and a consecutive wash-out phase of two weeks by chronic angiotensin II infusion or L-NAME application, respectively. We hypothesize that the changes in endothelial plasticity observed in WP2 following transient high salt intake are the basis for exacerbated vascular injury after AngII (400ng/ml/min) or L-NAME (20mg/kg/d) treatment. To test this, we will assess vascular function in vivo and ex vivo and analyze blood pressure as well as vascular immune cell infiltration. In addition, we will isolate ECs from the different arterial beds noted above and again perform bulkRNAseq and ATACseq analysis. This analysis will decipher whether the signaling pathways and transcriptome signatures identified in WP2 are the cause of the exaggerated hypertensive vascular damage. Within this approach, we will create the basis for upcoming projects treating the “salt memory” effect and thereby protecting from hypertensive vascular damage.
(